Entramos na
nona parte desta Linha do Tempo da
Química Medicinal e vamos tratar agora dos fármacos anti-virais que durante
muitos anos foram considerados ineficazes, quando pouco se conhecia sobre o
ciclo evolutivo destes microrganismos.
O início
desta história se dá por volta dos anos 50, do século passado, quando
identificou-se as propriedades biológicas da beta-tiossemicarbazona, uma das
primeiras substâncias ativas. Até então, os virologistas insistiam em avaliar o
eventual efeito anti-viral em antibióticos, então disponíveis. Obviamente que o
insucesso desta abordagem foi imenso, até que em 1957, Isaacs e Lindenmann
descrevem o interferon e seu efeito no vírus da hepatite-B. As viroses provocam
doenças severas e graves como varíola, poliomielite, influenza (e.g. gripe da ave), hepatite, AIDS entre
outras, e podem ser controladas de forma preventiva através de vacinas, em
alguns casos, nossa Linha do Tempo da
Química Medicinal não se ocupará desta vertente pois nossa vocação são
somente os fármacos.
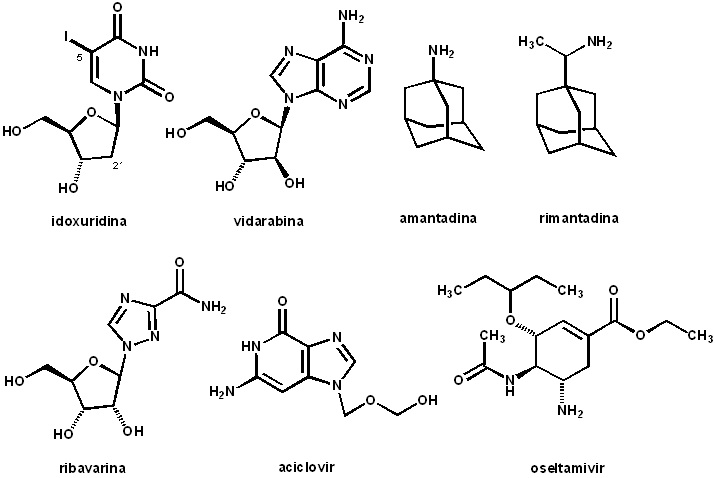
A
quimioterapia anti-viral começa em 1959, com o idoxuridine (IDU), que é o
5-iôdo-2’-deoxyuridina (Figura), análogo sintético de nucleosídeos sintetizado
nos laboratórios de William Herman Prusof (1920-2011) na Universidade de Yale,
EUA. Este fármaco descoberto pelo “pai da quimioterapia viral” e se mostrou
efetivo para o controle do vírus Herpes
simplex (HSV), sobretudo em infecções de pele. Considerado um potente
agente citotóxico, foi adotado para uso tópico, sendo empregado até hoje.
Alguns anos depois, em 1964, surge a vidarabina (Ara-A), anti-viral descoberto Stanford Research Institute, sintetizado
por Bernard R. Baker “copiando” a estrutura de dois nucleosídeos isolados de
uma esponja caribenha (Tethya crypta) que possuia uma sub-unidade arabinosídica
no lugar da clássica ribose. A vidarabina é uma substância nucleosídica
modificada, sendo um híbrido entre os nucleosídeos clássicos que contém ribose
e aqueles isolados do organismo marinho. A clássica ribose foi substituída pela
arabinose dos glicosídeos marinhos. Este fármaco antiviral, foi um dos
primeiros que pôde ser empregado sistemicamente devido ao seu reduzido perfil
tóxico. É ainda nesta época que surge a amantadina e uma molécula irmã, a rimantadine,
ambas possuindo um sistema biciclico adamantânico, que se mostraram eficazes no
tratamento da influenza, sendo um dos primeiros fármacos antivirais
não–nucleosídicos, atuando ao nível de proteínas virais envolvidas no processo
de replicação. O combate à influenza empregando estes fármacos permitiu
identificarem-se a capacidade de desenvolvimento da resistência viral. Inúmeros
estudos posteriores culminaram, em 1993, no surgimento dos fármacos antivirais
inibidores de neuroaminidase (NA), como veremos infra.
Em 1970,
Sidwell e colaboradores, trabalhando na empresa ICN Pharmaceuticals Inc. sintetizaram
a ribavirina, primeiro fármaco antiviral com amplo espectro de ação, ativo em
várias famílias de vírus RNA e DNA. Vale mencionar que não se identificou
desenvolvimento de resistência para este fármaco, indicando, provavelmente, o
envolvimento de mecanismo indireto para sua atividade.
A história
da origem dos fármacos anti-virais tem um capítulo muito significativo,
representado pela descoberta do aciclovir (ZoviraxR), realizada pela Dra
Gertrude Belle Elion (1918-1999), nos
laboratórios da Burroughs Welcome nos EUA e Welcome na Inglaterra. A
contribuição da Dra Elion à Química Medicinal foi imensa pois seu nome está
ligado a descoberta de vários fármacos: inter-alia
6-mercaptopurina para leucemia; azatioprina, imunosupressor; alopurinol, para o
trtamento da gôta; pirimetamina, para a malária; trimetoprim, para infecções
bacterianas). Por esta magnífica contribuição a Dra Elion foi indicada ao
prêmio Nobel de Medicina, em 1988, compartilhando-o com seu colega de
laboratório George H. Hitchings (1905-1998) e com Sir James W. Black (1924-2010), inventor do propranolol descrito na
parte V desta Linha do Tempo da Química
Medicinal. A descoberta do aciclovir marca o surgimento da segunda geração
de análogos de nucleosídeos, sendo este fármaco o mais seguro da classe, com
primeira indicação para do tratamento da infecção por HSV. Cabe mencionar que o
prefixo a, representa a
ausência do ciclo furanosídico da ribose do nucleosídeo, e a despeito desta
dissimilaridade molecular, este fármaco é fosforilado por timidino-quinase
viral resultando no mono-fosfato que por ação de outras quinases virais produz
o tri-fosfato de aciclovir, ativo sobre a DNA-polimerase do HSV.
Nos anos 80,
com a quimioterapia antiviral já bem estabelecida, surge um novo e tremendo
desafio, representado pela descoberta do vírus da síndrome da imunodeficiência
adquirida (SIDA = AIDS). Este vírus, da classe dos retrovírus (HIV) foi
descoberto em 1983 e descrito numa publicação na revista Science (DOI: 10.1126/science.6189183) por Luc Montagnier,
Françoise Barré-Sinouss e colaboradores,
do Instituto Pasteur, FR, o que valeu a estes pesquisadores o prêmio Nobel de
Medicina, em 2008. O primeiro fármaco antiviral descrito para a AIDS foi o AZT
(azidotimidina), derivado sintético da quimioteca da Burroughs Wellcome, Inglaterra, que foi bioensaiado contra o retrovírus
HIV no National Cancer Institute
(USA), e descrito em 1985 como inibidor efetivo de retroviruses. O mecanismo de
ação do AZT consiste numa fosforilação da hidroxila primária do fármaco por
quinases virais, levando ao trifosfato-AZT que é capaz de interagir e inibir a
enzima viral transcriptase-reversa (TR), atuando como um agente terminador de
cadeia. Após a descoberta do AZT inúmeros outros didesoxinucleosídeos, atuando
por este mecanismo foram descritos. Identificaram-se ao nível da TR um sítio
alostérico de interação que representaria um novo alvo-terapêutico possível para
o combate a AIDS. Após inúmeros esforços de pesquisa nesta área,
identificaram-se os anti-virais não-nucleosídicos inibidores de TR (NNRTIs = non-nucleoside reverse transcriptase
inhibitors) onde encontram-se nevirapina, delavirdina e efavirenz.

Estudos do
genoma viral indicaram a existência de protease vírus-específica que poderia,
também, representar um novo alvo-terapêutico para o tratamento da AIDS. Desta
feita, tratando-se de um mecanismo de ação distinto abria-se enormes
possibilidades representando possível tratamento de cêpas resistentes aos TR´i,
assim como novas associações e combinações terapêuticas de maior eficácia. Os
primeiros inibidores desta aspartato-protease viral surgem em 1995, representado
pelo saquinavir, precursor do indinavir (1996) e ritonavir (1996), a seguir
surgem o nelfinavir (1999) e amprenavir (1999) e por último o atazanavir
(2003). O combate ao HIV ganhou importante recurso quimioterápico representado
pelo maraviroc (2007), um novo agente anti-retroviral que atua ao nível de
citoquinas (CCR5) presentes nas membranas de células imunoativas humanas, que são
reconhecidas pelo vírus e facilitam sua internalização para as células do
hospedeiro. Inibindo esta citoquina, previne-se a internalização do vírus
prejudicando seu ciclo evolutivo celular.
A Linha do Tempo da Química Medicinal para
concluir esta parte relativa aos fármacos anti-virais não pode deixar de
mencionar aqueles que são eficazes no tratmento da gripe aviária (vírus H5N1;
H1N1, H3N2), atuando como inibidores de neuroaminidase (NA), proteína presente
no envelope viral cuja estrutura foi elucidada e teve seu mecanismo de ação
esclarecido, regulando a internalização viral, por interação com resíduos de
ácido sialíco de membrana de células plasmáticas do hospedeiro. Durante projeto
de descoberta racional de fármacos anti-virais, análogos modificados do ácido
siálico, levaram à identificação do zanamivir (1989; RelenzaR), primeiro
inibidor de NA anti-viral, comercializado pela GSK, a partir de 1993. Embora
eficiente, este fármaco, devido à sua baixíssima biodisponibilidade, é
empregado por via nasal o que estimulou estudos subsequentes visando a
identificação de NAi ativos por via oral. Estes estudos culminaram com a
descoberta do oseltamivir (TamifluR), em 2009, que é um pró-fármaco, empregado
na forma do precursor éster etílico, que por ação de esterases libera a forma
ativa do inibidor competitivo de NA.
Na próxima parte desta Linha do Tempo da Química Medicinal, a décima, trataremos da
descoberta ou invenção dos anti-inflamatórios não esteróides de segunda geração
(e.g. celecoxibe).
Obrigado por ler.
