Entramos na décima
segunda parte desta Linha do Tempo da
Química Medicinal e chegamos ao final do século XX, quando a quimioterapia
do câncer ganhou importantes fármacos, representados pelos inibidores de
tirosina-quinases (TK´s) que representam, quiça, a última inovação terapêutica
do século. São destes fármacos que vamos tratar nesta ocasião.
A
quimioterapia do câncer teve nos alcaloides bis-indólicos da Vinca (Catharanthus roseus (L.) G. Don.),
vincristina (VCR) e vimblastina (VCB), um dos seus primeiros recursos. Estes
fármacos, de origem natural, com propriedades antimitóticas, datam da década de
50 do século passado e foram objeto de estudos de diversos grupos de pesquisa,
tendo sido isolados por Ralph Noble e Charles Beer da Universidade de Western
Ontario, Canadá, que identificaram suas propriedades depressoras nas células
brancas sanguíneas. Isolados das folhas e flores de Vinca, pequenos, mas
vistosos arbustos, tiveram suas estruturas elucidadas pelo grupo de pesquisa do
Professor Ernest Wenkert, químico austríaco trabalhando na Universidade de San
Diego, EUA, mais ou menos na mesma época em que os primeiros aparelhos de
ressonância magnética nuclear de hidrogênio ficaram acessíveis. Claro que nesta
época em nada se comparavam a aqueles disponíveis nos dias de hoje, que os
superam em ca. 400 vezes, em potência
de campo. Estes produtos naturais foram inicialmente bioensaiados para o
tratamento da diabetes, entretanto mostraram-se capazes de produzirem severa
depressão na atividade da medula o que sugeriu sua eventual aplicação na
terapia do câncer. Estudos subsequentes conduzidos por Gordon Svoboda nos
laboratórios Eli Lilly, EUA, culminaram com seu lançamento na terapêutica em 1953,
com indicação para o tratamento de leucemias e linfomas, inclusive em crianças,
devido às propriedades antimitóticas e antitubulinícas destes compostos.

Pode-se dizer que é na quimioterapia do câncer que
encontramos um dos melhores exemplos da contribuição dos produtos naturais à
terapêutica. Em parte, graças a um ambicioso programa do National Cancer Institute dos EUA, iniciado em 1960, visando a
identificação de propriedades citotóxicas em diversas cepas celulares, de
extratos vegetais, inicialmente obtidos junto ao Departamento de Agricultura
norte-americano e posteriormente oriundos do mundo inteiro. As amostras eram
estudadas no Research Triangle Park, na Carolina do Norte, EUA. Esta busca de
novos padrões moleculares citotóxicos resultou alguns dos mais eficientes
fármacos anti-câncer que integram nosso arsenal terapêutico contemporâneo como
o paclitaxel, sesquiterpeno polifuncionalizado, de estrutura química inusitada,
isolado de Taxus brevifolia por Monroe
Wall e Mansukh Wani dos laboratórios do Triangle Research Park. A despeito de
sua complexa estrutura química, foram descritas sínteses totais para este
produto natural por Robert A Holton da Universidade da Florida e K C Nicolauo
do Scripps, California em 1994, além daquela relatada por Samuel J Danishefsky
da Universidade de Columbia, Nova Iorque, EUA, em 1996 e uma quarta por Paul A
Wender, em 1977. Para coroar os esforços do grupo de pesquisa do Research
Triangle Park, foi isolado, nos mesmos laboratórios, a camptotecina, alcaloide
quinolínico de biossíntese mixta, que caracterizou-se por apresentar em sua
estrutura pentaciclíca uma sub-unidade iridoidíca contida num anel alfa-hidróxilactona.
Vale mencionar que ambos os produtos naturais que se tornaram fármacos,
descobertos por Wall e Wani, possuíam estruturas químicas originais, com
padrões moleculares inéditos e apresentaram novos mecanismos farmacológicos de
ação, representando, em ambos os casos, autênticas inovações terapêuticas.
Enquanto que o paclitaxel (TaxolR) mostrou-se um inibidor de
tubulinas, a camptotecina, isolada de Campthoteca
acuminata, foi o primeiro fármaco anticâncer a atuar como inibidor de
topoisomerase-1. Além destas felizes coincidências, ambos compostos inspiraram
outras substâncias, sintéticas ou hemi-sintéticas, que ampliaram o impacto
terapêutico de suas descobertas. Desta forma o paclitaxel inspirou o docetaxel,
cabazitaxel e ortataxel, sendo este último ativo por via oral, enquanto que a
camptotecina levou aos derivados piridocarbazóis da classe dos tecanos (e.g. irinotecano e topotecano). Além
destes produtos naturais vegetais, outros como os derivados da podofilotoxina,
etoposido e tenoposido, completam os exemplos da importância dos produtos
naturais na quimioterapia do câncer. Não se deve deixar de mencionar os derivados
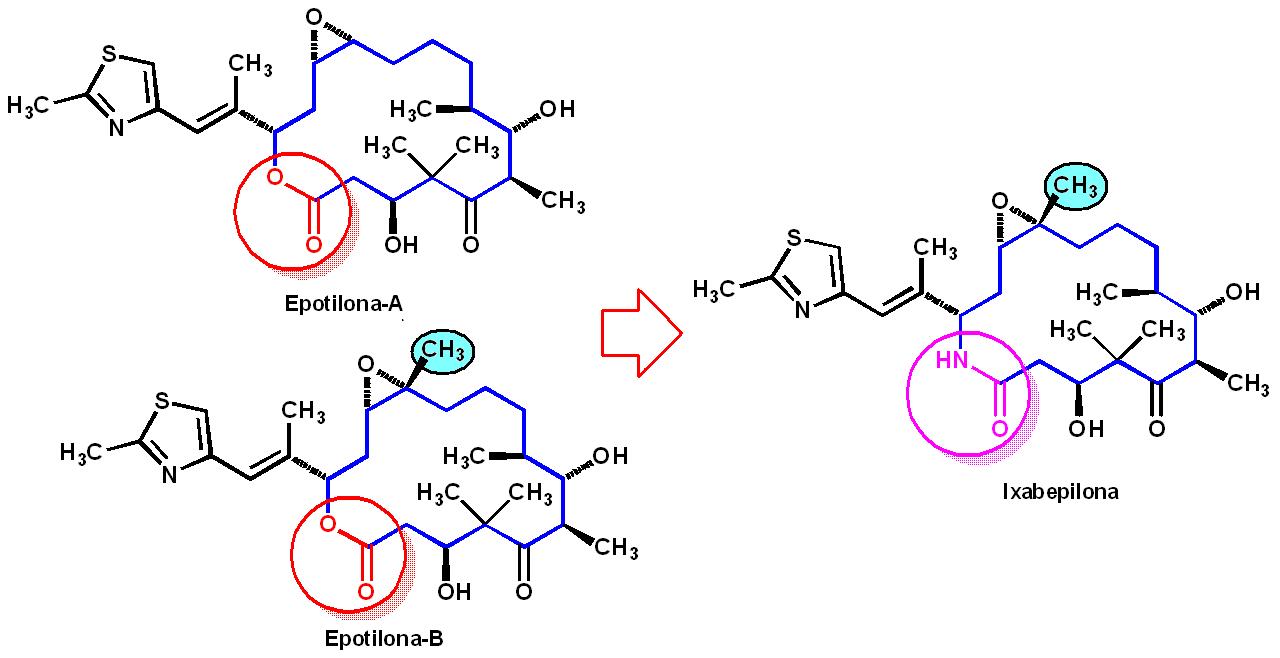
de origem microbiológica, representados por alguns antibióticos macrociclícos e
mais recentemente pelas epotilonas A e B, isoladas de Sorangium cellulosium. Estes últimos produtos naturais são lactonas
macrociclícas de 16 membros, que nas mãos dos químicos medicinais dos
laboratórios Bristol Meyers-Squibb (BMS) levaram a ixabepilona (IxempraR),
análogo macrociclíco que possui em substituição à função éster macrociclíco, de
fácil hidrólise enzimática na biofase, uma função bioisostérica macrolactama,
resistente às esterases, lançado no mercado em 2007.

A história dos
tinibes se inicia nos idos dos anos 70, quando as quinases despertaram o
interesse de inúmeros grupos de pesquisas, acadêmicos e industriais, como alvo
terapêutico possível, para o tratamento de várias doenças. Após os estudos do
genoma humano se identificam ca. 500
quinases. Estas enzimas representam importante família de enzimas
fosforilantes, utilisando o ATP como reagente de fosforilação, que modulam
importantes respostas celulares, ativando várias cascatas de sinalização. Em
1973, Janet Rowley e colaboradores determinam que a parte faltante do
cromossomo 22 – cromossomo Filadélfia - tinha sido translocada para o
cromossomo 9, em pacientes com leucemia mielóide crônica (CML). Em anos
seguintes, foi estabelecido que um gene de cada cromossomo (Bcr no cromossomo
22 e Abl no cromossomo 9), fundiam-se para formar um novo gene Bcr-ABl que
produzia uma proteína que provocava a CML. Deve-se aos trabalhos de David
Baltimore, Massachusetts, EUA, que identifica a natureza desta proteína como
sendo uma quinase. Alguns especialistas estimam que ca. 25% dos projetos de pesquisa em novos fármacos no mundo tenham
estas proteínas como alvos terapêuticos. Na classe das quinases encontram-se as
tirosinas-quinases (TKs), que promovem a fosforilação de proteínas na hidroxila
da tirosina, ativando mecanismos celulares que estimulam sua proliferação e
onde se enquadra a proteína caracterizada por Baltimore. Nos laboratórios
Ciba-Geigy (atual Novartis) iniciou-se, em 1982, um programa de pesquisas,
liderado por Jürg Zimmermann, visando identificarem-se um inibidor de quinases.
As empresas farmacêuticas que descobrem ou inventam fármacos, iniciavam, nesta
época, com muita expectativa, o uso de técnicas robotizadas de avaliação maciça
de amostras (high-throughput screening
- HTS), oriundas ou não de coleções de substâncias. Nestes estudos foram
identificados na Ciba-Geigy, derivados 2-fenilaminopiridinícos (em cor de rosa
na estrutura do imatinibe abaixo), capazes de inibirem a proteína-quinase C
(PKC) usada nestes ensaios in vitro.
A partir deste composto inicial, identificado neste screening, várias modificações moleculares foram sendo introduzidas,
visando potencializar seu efeito sobre outras quinases, dentre as quais estavam
a Abl-Bcr quinase, enzima da classe das TKs. Desta forma, os químicos
medicinais chegaram à estrutura do imatinibe (GlevecR), primeiro
inibidor de TK introduzido no mercado, em 2001 pela Novartis. Vale menção o
fato de que a introdução do grupo metila na posição orto do sistema diarilamina do imatinibe, foi um adereço estrutural
crítico para assegurar a conformação adequada à seletividade sobre a Abl-Bcr
TK. O sucesso do imatinibe, desenvolvido na Novartis, com perfil de
seletividade sobre TKs para o tratamento do câncer, representava um novo
mecanismo de controle desta doença e levou as empresas farmacêuticas
concorrentes a identificarem vários outros fármacos me-too com perfil inibidor multi-TK’s e com diferentes indicações
terapêuticas para outros tipos de câncer, como gefitinibe (IressaR
Astra-Zeneca), dasatinibe (SprycelR, BMS), sorafenibe (NexavarR,
Bayer e Onyx Pharmaceuticals), nilotinibe (TesignaR, Novartis),
lapatinibe (TykerbR, GSK), sunitibe (SutentR, Pfizer),
ruxolitinibe (JakafiR, Incyte Pharmaceuticals e Novartis) e crizotinibe
(XalkoriR, Pfizer), entre outros. A figura abaixo contém as
estruturas de alguns tinibes indicando por cores a similaridade molecular entre
estes fármacos.
O mesilato
de imatinibe foi o primeiro exemplo de sucesso da aplicação da técnica de HTS
de coleções de substâncias concomitante à técnicas de desenho molecular da Química
Medicinal, que representou uma importante inovação terapêutica.
Concluindo
esta parte estamos encerrando nossa Linha
do Tempo da Química Medicinal referente às descobertas ou invenções terapêuticas
do século XX e nas próximas partes passaremos às inovações ou descobertas deste
século, com mais de uma década vivida.
Obrigado por ler.